
Quality by Design for ANDAs: An Example for Immediate-Release Dosage Forms
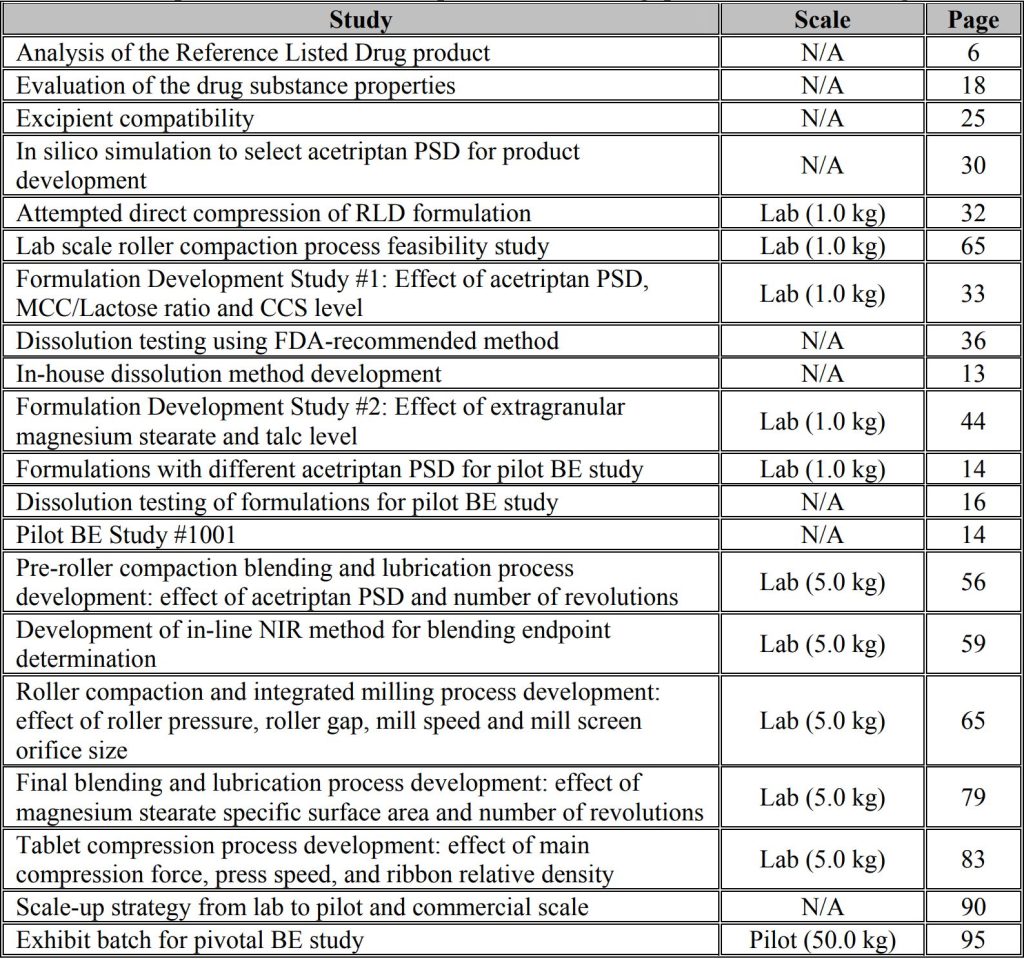
This is an example pharmaceutical development report illustrating how ANDA applicants can move toward implementation of Quality by Design (QbD). The purpose of the example is to illustrate the types of pharmaceutical development studies ANDA applicants may use as they implement QbD in their generic product development and to promote discussion on how OGD would use this information in review.
Although we have tried to make this example as realistic as possible, the development of a real product may differ from this example. The example is for illustrative purposes and, depending on applicants’ experience and knowledge, the degree of experimentation for a particular product may vary. The impact of experience and knowledge should be thoroughly explained in the submission. The risk assessment process is one avenue for this explanation. At many places in this example, alternative pharmaceutical development approaches would also be appropriate.
Notes to the reader are included in italics throughout the text. Questions and comments may be sent to [email protected]
Download the full document as PDF here: Quality by Design for ANDAs: An Example for Immediate-Release Dosage Forms
or read it here
April 2012
What is an ANDA?
ANDA stands for Abbreviated New Drug Application. It is a regulatory submission that a pharmaceutical company must file with the US Food and Drug Administration (FDA) to seek approval to market and sell a generic version of a drug that has already been approved by the FDA under a New Drug Application (NDA).
The ANDA pathway is a streamlined regulatory pathway designed to reduce the time and cost of drug development for generic drugs, while ensuring that they meet the same quality, safety, and efficacy standards as the original, brand-name drugs. Instead of conducting extensive clinical trials, as is required for an NDA, an ANDA relies on bioequivalence studies to demonstrate that the generic drug is equivalent to the reference listed drug, which is the original, brand-name drug that has been previously approved by the FDA under an NDA.
To be considered bioequivalent, the generic drug must have the same active ingredient, dosage form, strength, route of administration, and intended use as the reference listed drug. Additionally, the generic drug must be demonstrated to be absorbed into the bloodstream at the same rate and to the same extent as the reference listed drug.
Once an ANDA is approved by the FDA, the generic drug can be marketed and sold in the United States, usually at a lower cost than the brand-name drug.
What is a NDA?
NDA stands for New Drug Application. It is a regulatory submission that a pharmaceutical company must file with the US Food and Drug Administration (FDA) to seek approval to market and sell a new drug in the United States.
The NDA process is designed to ensure that new drugs are safe and effective for use in patients. The application requires extensive data from clinical trials to demonstrate the safety and efficacy of the drug, as well as information on the manufacturing process, labeling, and any potential risks or adverse effects associated with the drug.
To be considered for approval, a drug must demonstrate significant benefits over existing treatments or be the first drug to treat a particular condition. The clinical trials must also demonstrate that the drug is safe for use in patients and does not pose any significant risks or side effects.
Once an NDA is approved by the FDA, the drug can be marketed and sold in the United States. However, the FDA may require additional studies or monitoring to be conducted after approval to ensure the ongoing safety and efficacy of the drug.
Overall, the NDA process is lengthy and complex, and requires significant resources and investment from pharmaceutical companies. However, it is an important regulatory pathway to ensure that new drugs are safe and effective for patients, and can bring new treatments to market to address unmet medical needs.
What is a 505(2)?
The regulatory pathway referred to as 505(b)(2) is a specific type of new drug application (NDA) in the United States that allows a company to submit an NDA for a drug that relies, in part, on data from previously approved drugs.
The 505(b)(2) pathway is designed to streamline the drug development and approval process by allowing a company to leverage existing data to support the safety and efficacy of a new drug. This can include data from previously approved drugs, as well as data from literature, public databases, or other sources.
A 505(b)(2) application may be appropriate in cases where a company is developing a drug that is similar to an already approved drug but has some differences, such as a new indication, dosage form, or route of administration. By leveraging existing data, a company may be able to reduce the time and cost associated with clinical trials, while still providing the FDA with the data necessary to evaluate the safety and efficacy of the drug.
However, it’s important to note that the 505(b)(2) pathway is not a shortcut to approval, and companies still need to meet the same regulatory standards for safety and efficacy as they would for a traditional NDA. Additionally, the FDA may require additional studies or data beyond what is already available to support the approval of a drug under the 505(b)(2) pathway.